
SHREC 2022: Protein-ligand binding site recognition
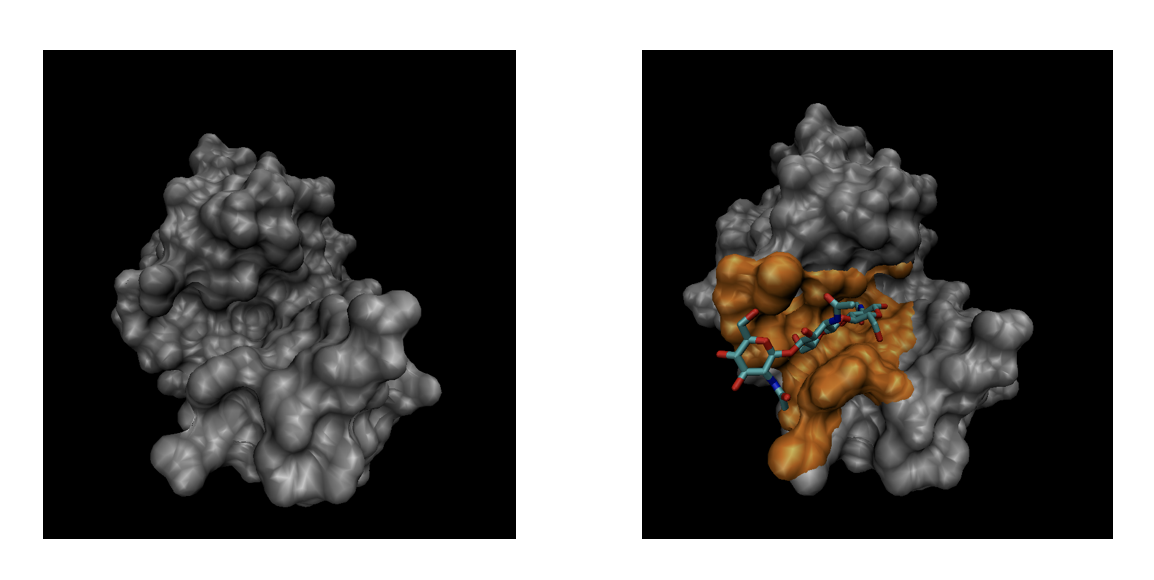
The general objective of this SHREC track is to evaluate the effectiveness of computational methods in recognizing ligand binding sites in a protein, based on the description of its molecular surface. Starting from a set of protein-ligand complex structures obtained via X-ray crystallography and deposited in the PDB repository, we build the proteins’ Solvent Excluded Surface (SES) by the freely available software NanoShaper. For each of these surfaces, we then identify the set of vertices (which we will refer to as segments) which are within 4 Å of any ligand atom. Additionally, for each surface we provide an anonymized PQR file, where it is possible to retrieve the atomic centers and radii; atoms within 5 Å of any ligand atom are flagged as determining a ligand binding site. We hereby refer to this set of vertices and atoms as binding pockets.
The track is jointly organized by IMATI-CNR and the CONCEPT lab at IIT. The final report of this SHREC track will be submitted as a joint contribution to the international journal Computers & Graphics and will follow a two-stage review process. The paper will be authored by the track coordinators and all participants who submitted their results.
Motivation
The molecular surface of a protein, i.e. the protein boundary not accessible to the solvent (water), plays a fundametal role in the characterization and prediction of the interactions of a protein with other proteins or ligands (small molecules).
In this study, we aim to identify pockets that could bind ligands, based on their shape.
The prediction of protein-ligand binding region is one of the focal points of activity in computational biophysics and structural biology. Indeed, when a small molecule binds to a protein it affects its biological behavior. The identification of candidate binding sites is therefore preliminary to molecular docking, which is a key tool for drug design. Adequate computational techniques to model these interactions are important because can leverage the information obtained from the growing number of known protein-ligands systems.
Description of the track
A dataset of approximately 1100 protein surfaces and corresponding to about 1750 relevant subregions (regions close to the ligand) will be provided to the participants. The dataset will be split into a training and a test set (in the proportion 85-15).
Each model will be represented by a file containing the protein structure molecular surface in OFF format and an anonymized PQR file. Moreover, for each structure of the training set the ligand binding sites will be collected in a TXT file (for the OFF file), and a classification vector (for the PQR file). The TXT file represents the ligand binding sites of the associated structure and it contains a vector, whose entries correspond to the vertices of the corresponding OFF file (in the same order); each vector entry is either zero (not known to contribute to a binding site) or an integer (different numbers identify distinct protein-ligand binding sites). The same information will be replicated in the one-but-last , i.e. the charge, column of the PQR file.
To compare the performance of the proposed methods we will ask the participants to provide us, for each protein in the test set, a vector representing the 10 most likely binding sites they have identified in the model, either in terms of the vertices (if using the OFF files) or of the atoms (if using the PQR files); we further ask the participants to provide an ordering on the 10 predicted binding sites, from the most to least likely. IMPORTANT NOTICE: i) A single structure can contain more than one binding sites. ii) The training set does not imply any ranking. All provided pockets are positive examples and should be considered equally important.
Performance Evaluation: The performance of the segmentation algorithms will be assessed by using structural evaluation measures (e.g., identification of the protein residues composing the observed binding sites by measuring the distance from the ligand in the crystal structure) and statistics (e.g., classification measures).
Registration and Other Procedures
Each participant is requested to register to the track by sending an email to Luca Gagliardi (email: luca.gagliardi@iit.it), Andrea Raffo (email: andrea.raffo@ge.imati.cnr.it) and Ulderico Fugacci (email: ulderico.fugacci@ge.imati.cnr.it) with the subject "SHREC 2022: Protein-ligand binding site recognition".
Then, an answer will be sent to each participant with further instructions.
We plan to submit the benchmark code and data to the replicability stamp
Further Information
- The evaluations will be done automatically;
- The track results will be combined into a joint paper to be submitted to Computers & Graphics;
- The description of the track and its results will be presented at the Symposium on 3D Object Retrieval 2022 (September, 2022).
Important dates
- Jan 3, 2022: the call for participantion is circulated.
- Jan 14, 2022: the training set is published; the participants start running their methods.
- Jan 25, 2022: the test set is published.
- Jan 25, 2022 -- Feb 14, 2022: the whole dataset is available for download.
- Feb 14, 2022: each participant provides the results of their run, the program (binary code) that generated the results and a summary of the proposed method.
- Feb 21, 2022: the outcome of the track is circulated among the participants; the participants comment the results.
- Feb 28, 2022: the organizers circulate a draft of track paper for feedback.
- Mar 15, 2022: the track paper is submitted for review.
- March - June, 2022: review of the track paper.
- September, 2022: workshop presentation.